“批文清查三部曲”之一:上市后再评价的未来棋局
发布时间:2017-03-03 20:55:01
医药网8月23日讯 药物1致性评价、工艺核对、上市后再评价被业内称为“批文清查3部曲”。除“1致性评价”是相对新鲜名词,后面二者多年来1直在进行,其中“上市后再评价”仍有后着。 6月30日,CFDA办公厅发布关于马来酸桂哌齐特注射液上市后临床研究有关事宜的通知,要求已获批的马来酸桂哌齐特注射液展开上市后临床研究,并要求其须在2018年6月30日前完成。 药品的安全性、有效性和质量可控愈来愈遭到我国药监部门的重视,已上市药品将面临上市后再评价的挑战。 思考:再评价政策对批文的影响 政策回顾与重要时间节点 上市后再评价研究的推动主要是为了保证药品的质量,1990年原卫生部药政局曾对西药地方标准品种进行再评价工作,并于1996年和1998年进1步明确了该项工作的范围、计划、原则和要求。1998年10月、11月,原国家局依照“整理1类,清算1类”的要求,经过国家药典委员会组织对非抗生素类抗感染药、解热镇痛药共400余种地方标准品种进行了再评价。由此可知,对已上市的药品而言,再评价研究其实不算新鲜事。 最近1次由原国家局发起的再评价是2009年,主要是加强中药注射剂的不良反应监测,并对重点品种如双黄连注射剂、参麦注射剂、鱼腥草注射液和鱼金注射液组织展开综合评价。2010年原国家局组织制定了《中药注射剂安全性再评价生产工艺评价技术原则(试行)》《中药注射剂安全性再评价质量控制评价技术原则(试行)》《中药注射剂安全性再评价非临床研究评价技术原则(试行)》《中药注射剂安全性再评价临床研究评价技术原则(试行)》《企业对中药注射剂风险控制能力评价技术原则(试行)》《中药注射剂安全性再评价风险效益评价技术原则(试行)》和《中药注射剂风险管理计划指点原则(试行)》共7个技术。 再评价对批文的淘汰作用 2002年3月21日前,原国家局曾对国内药品生产企业所有合法生产的药品批准文号进行换发,包括各药品生产企业历年经药品监管部门批准的合法生产的所有药品(含生物制品,背规审批的药品除外)。当时参加化学药品再评价的品种和中成药地方标准品种和中药保健药品,在完成整理工作后也1并换发了新的药品批准文号。 2009年,原国家局结合药品批准文号清查工作展开药品再注册。药品再注册批准证明文件自签发之日起生效,有效期为5年。不具有生产条件、质量不能保证、安全风险高的品种将会不予再注册。当时清除1批注射剂类药品,即生产工艺和处方核对工作中未核对的、或核对结果为“责令停产”的药品。 通过分析2002年的批文,2009⑵010年的再注册批文和5年1次再注册批文的现状,可以从侧面了解再评价政策的影响程度。 那末,中药注射剂的再评价对批文的注销真的起到推动作用了吗? 咸达数据V3.2发现,参麦注射液普遍都在2015年再注册成功,从批文的注册来看,暂无批文是由于再评价研究致使批文注销。 双黄连注射剂方面,截至2016年8月15日,共有批文15个。其中,产品名为“注射用双黄连(含冻干)”的批文4个,分别来自黑龙江省松花江药业有限公司和哈药团体中药2厂两个厂家;产品名为“双黄连注射液”相对批文多1些,该产品名共有11个批文。相对2002年的26个批文[双黄连注射液21个批文,注射用双黄连(含冻干)5个批文],批文数是有所减少的。对减少的批文进1步分析后发现,被注销的批文大多是在2010年前后没有再注册的批文。经过2010年的再注册近期没有再注册的批文只有1个,为黑龙江省松花江药业有限公司的国药准字Z20063433。 鱼腥草注射液从生产批文来看,现在还有108个批文,较原本的196个批文的确减少了很多,但是大部份注销的批文都因没有通过2010年前后的再注册。通过2010年再注册后2015年没有再注册的批件唯一5个,分别是国药准字Z52020341、国药准字Z52020342、国药准字Z52020343、国药准字Z52020512和国药准字Z52020513。 综上所述,2009年所推动的再评价研究影响了双黄连注射剂和鱼腥草注射液的再注册。2009⑵010年间已通过再注册的批文,尔后再被注销的批文就不多了。 中药独家品种再评价动力足 从CDE公然的数据库可以看到,2013年以来,在进行的上市后再评价的中药产品(有在CDE登记公然的)仅悦康银杏叶提取物注射液、参泽舒肝胶囊和麝香通心滴丸。其中,银杏叶提取物注射液和参泽舒肝胶囊主要再评价安全性,麝香通心滴丸主要针对其医治冠心病稳定型心绞痛的疗效。在进行Ⅳ期临床实验的有龙血通络胶囊、双冬胶囊、金香疏肝片、和胃止泻胶囊和大黄总蒽醌胶囊。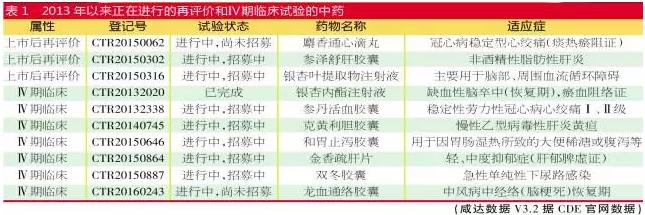 以上数据表明,2010年以后上市的独家中药口服药大多会启动上市后再评价项目。相对而言,2002年上市的中药注射剂由于生产家数多,竞争大,因此没有多大动力去履行上市再评价项目。 趋势:未来的几个重要政策导向 药品上市后再评价是1个长时间、延续的进程。虽然依照现有的药事管理制度,监管部门在药品上市前的研发、审批阶段已对该药品的风险与收益平衡机制做了评估,但出于药品的安全性等需求,药品上市后仍需要继续评价。而药品的不良反应有可能随着用量的增加逐步被暴露。另外,上市多年的“老药”也要监测其与其他药品联合使用时出现新不良反应的可能性。 美日经验:他山之石的鉴戒与启发 先来看看美国经验。美国通过药物监察项目搜集报告,建立药品不良事件报告系统数据库;评估药品商品名、标签、包装,避免处方、分发和药品管理中出现的医治毛病,以免因药品商品名与已上市的其他产品类似而产生的使用毛病。另外,美国药品评价与研究中心还建立了“药品再评价质量管理规范”,该规范包括对药品审评的进程、程序、内容、管理方式等诸多方面的规定,不单单局限于上市后药品再评价工作。 日本则将上市后药品再评价工作分为“定期再评价”和“临时再评价”两种模式。“定期再评价”是通过国内外文献调研做出是不是进行再评价的挑选,在挑选进程中,会挑选出有问题的文献,并对问题文献进行综合整理,拜托企业调查。受拜托企业提交《文献调查报告书》后,由“药事食品卫生审议会”肯定是不是进行再评价。“临时再评价”是针对紧急情况采取的评价措施。 我国政策:关联药典淘汰机制,未来纳入再评价的方向 为何新获批的中药新药需要启动上市后再评价?主要是为了让药品进入国家基本药物目录、医疗保险目录遴选,提供上市后再评价的临床研究数据将有助于厂家证明其药物的临床地位。 2016年2月14日,国务院召开常务会议,部署推动医药产业创新升级,会议肯定加大中医药投入和政策扶持,在国家基本药物目录中增加中成药品种数量。中药的政策利好,更是推动了独家中药增加再评价研究的投入。 2020年版《中国药典》近期亦开始启动编辑。2020版药典将以临床价值为导向,重点关注临床用量大、安全风险高、质量问题较多的品种,结合国家药品标准清算,建立标准淘汰机制。1是对现有国家药品标准进行清算,不符合药典收载原则的,药典将不再予以收载;2是淘汰那些没有文号、长时间没有生产、没法控制质量的标准;3是需要对药品临床价值或风险效益进行评价的,药品再评价研究数据不佳的将要面临淘汰。这亦推动企业展开再评价项目。 从安全性的角度而言,国家下1步将有可能要求多组分生化药注射剂、中药口服药和中药配方颗粒进行上市再评价研究。中药注射剂的上市再评价有可能像化药1致性评价那样设定最后期限。部份安全性和疗效存疑、临床用量较大的药品也可能进入国家再评价研究名单。 另外,国家要求改进型药品需要证明其临床有效性,而按旧化学分类获批的改进型药品基本没有做相干的实验研究。改进型药品不适用1致性评价实验,因此政策面将有可能鼓励相干药品在1定期限内展开再评价实验。 而对国外已上市使用但国内缺少且临床急需的儿童用药品种,目前儿科临床实验的履行难度较大,未来也有可能放开注册的限制或下降临床实验门坎,但须对上市后的儿童用药进行再评价,以最大程度保障儿童用药安全,并为儿童科学、公道用药提供信息支持。
以上数据表明,2010年以后上市的独家中药口服药大多会启动上市后再评价项目。相对而言,2002年上市的中药注射剂由于生产家数多,竞争大,因此没有多大动力去履行上市再评价项目。 趋势:未来的几个重要政策导向 药品上市后再评价是1个长时间、延续的进程。虽然依照现有的药事管理制度,监管部门在药品上市前的研发、审批阶段已对该药品的风险与收益平衡机制做了评估,但出于药品的安全性等需求,药品上市后仍需要继续评价。而药品的不良反应有可能随着用量的增加逐步被暴露。另外,上市多年的“老药”也要监测其与其他药品联合使用时出现新不良反应的可能性。 美日经验:他山之石的鉴戒与启发 先来看看美国经验。美国通过药物监察项目搜集报告,建立药品不良事件报告系统数据库;评估药品商品名、标签、包装,避免处方、分发和药品管理中出现的医治毛病,以免因药品商品名与已上市的其他产品类似而产生的使用毛病。另外,美国药品评价与研究中心还建立了“药品再评价质量管理规范”,该规范包括对药品审评的进程、程序、内容、管理方式等诸多方面的规定,不单单局限于上市后药品再评价工作。 日本则将上市后药品再评价工作分为“定期再评价”和“临时再评价”两种模式。“定期再评价”是通过国内外文献调研做出是不是进行再评价的挑选,在挑选进程中,会挑选出有问题的文献,并对问题文献进行综合整理,拜托企业调查。受拜托企业提交《文献调查报告书》后,由“药事食品卫生审议会”肯定是不是进行再评价。“临时再评价”是针对紧急情况采取的评价措施。 我国政策:关联药典淘汰机制,未来纳入再评价的方向 为何新获批的中药新药需要启动上市后再评价?主要是为了让药品进入国家基本药物目录、医疗保险目录遴选,提供上市后再评价的临床研究数据将有助于厂家证明其药物的临床地位。 2016年2月14日,国务院召开常务会议,部署推动医药产业创新升级,会议肯定加大中医药投入和政策扶持,在国家基本药物目录中增加中成药品种数量。中药的政策利好,更是推动了独家中药增加再评价研究的投入。 2020年版《中国药典》近期亦开始启动编辑。2020版药典将以临床价值为导向,重点关注临床用量大、安全风险高、质量问题较多的品种,结合国家药品标准清算,建立标准淘汰机制。1是对现有国家药品标准进行清算,不符合药典收载原则的,药典将不再予以收载;2是淘汰那些没有文号、长时间没有生产、没法控制质量的标准;3是需要对药品临床价值或风险效益进行评价的,药品再评价研究数据不佳的将要面临淘汰。这亦推动企业展开再评价项目。 从安全性的角度而言,国家下1步将有可能要求多组分生化药注射剂、中药口服药和中药配方颗粒进行上市再评价研究。中药注射剂的上市再评价有可能像化药1致性评价那样设定最后期限。部份安全性和疗效存疑、临床用量较大的药品也可能进入国家再评价研究名单。 另外,国家要求改进型药品需要证明其临床有效性,而按旧化学分类获批的改进型药品基本没有做相干的实验研究。改进型药品不适用1致性评价实验,因此政策面将有可能鼓励相干药品在1定期限内展开再评价实验。 而对国外已上市使用但国内缺少且临床急需的儿童用药品种,目前儿科临床实验的履行难度较大,未来也有可能放开注册的限制或下降临床实验门坎,但须对上市后的儿童用药进行再评价,以最大程度保障儿童用药安全,并为儿童科学、公道用药提供信息支持。










