
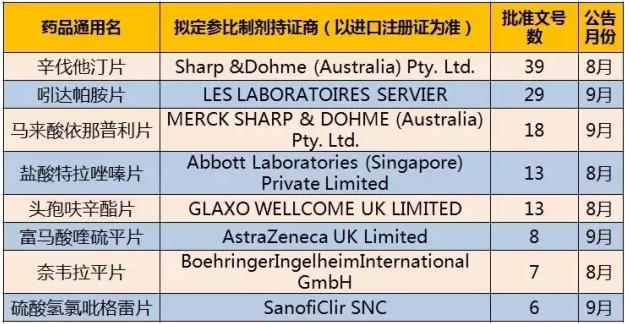
 (数据来源:咸达数据V3.2) 企业提交的参比制剂备案信息可以看出哪些产品是企业重点关注的。从中检院提供的企业参比试剂备案可以看出,截至2016年6月31日,共942个厂家产品参与了备案,其中509个厂家产品在289个仿造药目录内。 咸达数据V3.2发现,本次中检院942个品种中,阿莫西林胶囊是最多企业备案的产品,其次是辛伐他汀片,第3是苯磺酸氨氯地平片。 以辛伐他汀片为例,比较中检院和企业备案的情况后可以发现,企业备案和中检院都锁定参比试剂的公司为默沙东,但企业备案对原研药生产厂家的英文名并没有统1名称,较难统计。 表2 企业提交的参比制剂备案数最多的产品名排名
(数据来源:咸达数据V3.2) 企业提交的参比制剂备案信息可以看出哪些产品是企业重点关注的。从中检院提供的企业参比试剂备案可以看出,截至2016年6月31日,共942个厂家产品参与了备案,其中509个厂家产品在289个仿造药目录内。 咸达数据V3.2发现,本次中检院942个品种中,阿莫西林胶囊是最多企业备案的产品,其次是辛伐他汀片,第3是苯磺酸氨氯地平片。 以辛伐他汀片为例,比较中检院和企业备案的情况后可以发现,企业备案和中检院都锁定参比试剂的公司为默沙东,但企业备案对原研药生产厂家的英文名并没有统1名称,较难统计。 表2 企业提交的参比制剂备案数最多的产品名排名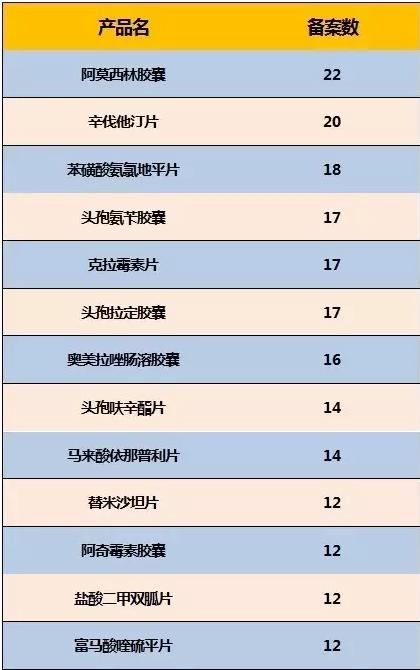 (数据来源:咸达数据V3.2) 根据中检院的数据,289目录产品有138个产品共509个品规已有企业备案原研,这意味着有将近50%的产品通用名目前企业已有初步研究意向。其中,阿莫西林胶囊和苯磺酸氨氯地平片是289目录中最多备案的产品,各有18个品规已备案。 咸达数据V3.2将企业备案的药品数对照CFDA于8月17日所发布的2018年底前须仿造药质量和疗效完成1致性评价品种批准文号数,发现头孢呋辛酯胶囊剂与片剂、阿卡波糖胶囊、复方利血平氨苯蝶啶片、虎魄酸亚铁片、环孢素胶囊、阿法骨化醇片、富马酸比索洛尔片和阿卡波糖片目前企业的备案数已同等目标完成1致性评价品种批准文号数,其中阿卡波糖胶囊、复方利血平氨苯蝶啶片、虎魄酸亚铁片和环孢素胶囊,都是只有1个批准文号需要做实验的药品。 942个企业备案数据中,备案最多的是山东新华制药股分有限公司,其次是石药团体欧意药业有限公司,第3是山东京卫制药有限公司。而2018年必须完成的289目录品种中,备案最多的企业是山东新华制药股分有限公司,其次是石药团体欧意药业有限公司,第3是华润双鹤利民药业(济南)有限公司。 小结 从数据来看,需要2018年完成1致性评价的基药口服制剂17,740个标准文号数唯一509个备案,不到3%,这意味着许多厂家还是在观望。所幸的是,289目录通用名中有138个药品有备案记录,亦即48%个药品已有厂家成心向观望,其中在289目录中只有1家申报的已备案药品共有45个,包括批文号数较多的阿司匹林肠溶片和双氯芬酸钠肠溶片。 北京近日对289目录的产品有高达300万元的资金支持,这将会鼓励北京企业对289目录药品1致性评价的参与度。 利好2:“改规格药品评价1般斟酌”政策发布 9月13日,CFDA公然征求仿造药质量和疗效1致性评价改规格药品评价1般斟酌的意见。这意味着此前大家1直争议的改规格药品的1致性评价该怎样做有了新的补充。 CFDA对改规格药品有了新的定义,改规格药品指该规格在欧盟、美国或日本均未获准上市或虽获准上市但没法肯定同规格参比制剂的药品,包括原研药品相应规格曾获批但已不存在或原研药品从未获批该规格的品种。 值得注意的是,CFDA今年发布的化学药品新注册分类中,要求仿造药必须与原研药具有相同活性成份、相同剂型、相同给药途径、相同规格;对改进后增加规格的,需按新药受理。改规格药品的评价包括药学和临床实验等多个方面的内容。 改规格药品需充分论证改规格药品存在的科学性、公道性和必要性,并且药品规格的变更应在其临床使用的用法用量范围内,亦即其规格应在单次最小给药剂量与单次最大给药剂量的范围内。 药学研究一样是改规格药品重点研究的内容,包括处方组成与工艺研究、质量标准与质量控制,和增加规格产品的稳定性实验研究。 实验要求方面,满足以下条件者,可选择原研同品种其他规格为参比制剂,以相同剂量给药进行人体生物等效性实验:仿造药和原研药的适应症和用法用量相同;在医治剂量范围内,药物显现线性药代动力学特点;在医治剂量范围内,药物显现线性药代动力学特点;改规格制剂与参比制剂的活性组分1致,且制剂处方比例类似;改规格制剂和参比制剂体外溶出、释放特点类似。不满足上述条件的改规格药品,CFDA建议采取临床实验进行评价。 小结 适用于1致性评价的改规格药品有了新的指点补充文件,对此类药品有了新的定义和相干的研究准则,这对相干企业是利好。 但是,对改进型改剂型的企业而言,目前还没有相干的文件发布。预计改剂型药品的疗效再评价的项目还是要做临床实验。 利好3:参比制剂找不到?优先选择安慰剂对比 9月14日,总局办公厅公然征求仿造药质量和疗效1致性评价临床有效性实验1般斟酌的意见。这是主要针对找不到参比制剂的仿造药提供临床有效性实验如何进行的补充指点。 对此类找不到参比制剂且国内已上市的仿造药,在进行临床有效性实验前应回顾并评估该药品在现有医治中的临床价值,并基于其国内外临床研究及其利用情况(如指南的地位,目前循证医学证据的情况),对该药品的临床有效性进行初步判断。生产厂家应斟酌该药物的临床疗效情况,与其他医治药物的疗效比较情况,和是不是存在影响现有医治药物疗效的其他因素(如耐受性、允从性或患者偏向性)。 为了证明药品的有效性,CFDA鼓励尽量选择安慰剂对比进行临床有效性实验,除细胞毒类药品等特殊情况不合适利用安慰剂对比而必须选择阳性对比药的情况。 另外,阳性对比药应适应症相同且最好机理相同,临床疗效确切且可预期,并且是公认的、有良好循证医学证据。不过,CFDA没有硬性规定阳性对比实验,从而相对下降了有效性实验的难度。 仿造药质量和疗效1致性评价临床有效性实验的比较类型主要包括优效性实验和非劣效性实验。 优效性实验的目的是显示实验药的医治效果优于对比药,非劣效性实验的目的是确证实验药的疗效最少不差于阳性对比药。CFDA鼓励生产厂家尽量选择优效性实验设计而没有硬性要求非劣性实验,从实验设计上又1次下降了临床有效性的难度。 CFDA认为,作为已上市仿造药,其疗效和安全性循证医学证据已有1定积累,临床终点可使用有价值的替换终点或生物标记物,以科学、准确、灵敏、有效地实现实验目的。这意味着此类仿造药无需像新药临床实验那般严格考核临床终点。 小结 这对找不到参比制剂的且具有临床疗效的仿造药是个利好,特别是曾在海外上市但现在退市的原研药及其改剂型的药品。在临床从严的背景下,虽然临床实验的难度有所下降,但是安全无效的药品仍难以取得更优的指向性结果。
(数据来源:咸达数据V3.2) 根据中检院的数据,289目录产品有138个产品共509个品规已有企业备案原研,这意味着有将近50%的产品通用名目前企业已有初步研究意向。其中,阿莫西林胶囊和苯磺酸氨氯地平片是289目录中最多备案的产品,各有18个品规已备案。 咸达数据V3.2将企业备案的药品数对照CFDA于8月17日所发布的2018年底前须仿造药质量和疗效完成1致性评价品种批准文号数,发现头孢呋辛酯胶囊剂与片剂、阿卡波糖胶囊、复方利血平氨苯蝶啶片、虎魄酸亚铁片、环孢素胶囊、阿法骨化醇片、富马酸比索洛尔片和阿卡波糖片目前企业的备案数已同等目标完成1致性评价品种批准文号数,其中阿卡波糖胶囊、复方利血平氨苯蝶啶片、虎魄酸亚铁片和环孢素胶囊,都是只有1个批准文号需要做实验的药品。 942个企业备案数据中,备案最多的是山东新华制药股分有限公司,其次是石药团体欧意药业有限公司,第3是山东京卫制药有限公司。而2018年必须完成的289目录品种中,备案最多的企业是山东新华制药股分有限公司,其次是石药团体欧意药业有限公司,第3是华润双鹤利民药业(济南)有限公司。 小结 从数据来看,需要2018年完成1致性评价的基药口服制剂17,740个标准文号数唯一509个备案,不到3%,这意味着许多厂家还是在观望。所幸的是,289目录通用名中有138个药品有备案记录,亦即48%个药品已有厂家成心向观望,其中在289目录中只有1家申报的已备案药品共有45个,包括批文号数较多的阿司匹林肠溶片和双氯芬酸钠肠溶片。 北京近日对289目录的产品有高达300万元的资金支持,这将会鼓励北京企业对289目录药品1致性评价的参与度。 利好2:“改规格药品评价1般斟酌”政策发布 9月13日,CFDA公然征求仿造药质量和疗效1致性评价改规格药品评价1般斟酌的意见。这意味着此前大家1直争议的改规格药品的1致性评价该怎样做有了新的补充。 CFDA对改规格药品有了新的定义,改规格药品指该规格在欧盟、美国或日本均未获准上市或虽获准上市但没法肯定同规格参比制剂的药品,包括原研药品相应规格曾获批但已不存在或原研药品从未获批该规格的品种。 值得注意的是,CFDA今年发布的化学药品新注册分类中,要求仿造药必须与原研药具有相同活性成份、相同剂型、相同给药途径、相同规格;对改进后增加规格的,需按新药受理。改规格药品的评价包括药学和临床实验等多个方面的内容。 改规格药品需充分论证改规格药品存在的科学性、公道性和必要性,并且药品规格的变更应在其临床使用的用法用量范围内,亦即其规格应在单次最小给药剂量与单次最大给药剂量的范围内。 药学研究一样是改规格药品重点研究的内容,包括处方组成与工艺研究、质量标准与质量控制,和增加规格产品的稳定性实验研究。 实验要求方面,满足以下条件者,可选择原研同品种其他规格为参比制剂,以相同剂量给药进行人体生物等效性实验:仿造药和原研药的适应症和用法用量相同;在医治剂量范围内,药物显现线性药代动力学特点;在医治剂量范围内,药物显现线性药代动力学特点;改规格制剂与参比制剂的活性组分1致,且制剂处方比例类似;改规格制剂和参比制剂体外溶出、释放特点类似。不满足上述条件的改规格药品,CFDA建议采取临床实验进行评价。 小结 适用于1致性评价的改规格药品有了新的指点补充文件,对此类药品有了新的定义和相干的研究准则,这对相干企业是利好。 但是,对改进型改剂型的企业而言,目前还没有相干的文件发布。预计改剂型药品的疗效再评价的项目还是要做临床实验。 利好3:参比制剂找不到?优先选择安慰剂对比 9月14日,总局办公厅公然征求仿造药质量和疗效1致性评价临床有效性实验1般斟酌的意见。这是主要针对找不到参比制剂的仿造药提供临床有效性实验如何进行的补充指点。 对此类找不到参比制剂且国内已上市的仿造药,在进行临床有效性实验前应回顾并评估该药品在现有医治中的临床价值,并基于其国内外临床研究及其利用情况(如指南的地位,目前循证医学证据的情况),对该药品的临床有效性进行初步判断。生产厂家应斟酌该药物的临床疗效情况,与其他医治药物的疗效比较情况,和是不是存在影响现有医治药物疗效的其他因素(如耐受性、允从性或患者偏向性)。 为了证明药品的有效性,CFDA鼓励尽量选择安慰剂对比进行临床有效性实验,除细胞毒类药品等特殊情况不合适利用安慰剂对比而必须选择阳性对比药的情况。 另外,阳性对比药应适应症相同且最好机理相同,临床疗效确切且可预期,并且是公认的、有良好循证医学证据。不过,CFDA没有硬性规定阳性对比实验,从而相对下降了有效性实验的难度。 仿造药质量和疗效1致性评价临床有效性实验的比较类型主要包括优效性实验和非劣效性实验。 优效性实验的目的是显示实验药的医治效果优于对比药,非劣效性实验的目的是确证实验药的疗效最少不差于阳性对比药。CFDA鼓励生产厂家尽量选择优效性实验设计而没有硬性要求非劣性实验,从实验设计上又1次下降了临床有效性的难度。 CFDA认为,作为已上市仿造药,其疗效和安全性循证医学证据已有1定积累,临床终点可使用有价值的替换终点或生物标记物,以科学、准确、灵敏、有效地实现实验目的。这意味着此类仿造药无需像新药临床实验那般严格考核临床终点。 小结 这对找不到参比制剂的且具有临床疗效的仿造药是个利好,特别是曾在海外上市但现在退市的原研药及其改剂型的药品。在临床从严的背景下,虽然临床实验的难度有所下降,但是安全无效的药品仍难以取得更优的指向性结果。









